Introduzione
La Retinite pigmentosa (RP) comprende un gruppo di distrofie retiniche ereditarie caratterizzate dalla degenerazione dei coni, che rappresentano i fotorecettori retinici.
La RP si caratterizza dalla perdita dei fotorecettori prima dei bastoncelli, seguiti poi dalla perdita anche dei coni.
I sintomi si associano a visione notturna ridotta la quale viene seguita dalla progressiva perdita del campo visivo fino ad arrivare ad una visione di tipo concentrica, che spesso esita fino alla cecità. La funzione visiva nella regione maculare è generalmente preservata fino agli stadi tardivi della malattia. Il reperto tipico di questa patologia si riscontra all’esame del fondo oculare mediante il riscontro di tipiche spicole ossee pigmentate situate prevalentemente in periferia e media periferia, che si associano ad attenuazione dei vasi retinici e pallore del disco ottico.
A livello clinico l’elettroretinogramma come indagine strumentale può essere dirimente nella diagnosi per rilevare la caratteristica perdita della funzione fotorecettoriale, prima a livello dei bastoncelli e successivamente tale perdita si estende anche ai coni.
La diagnosi deve essere sempre confermata da un elettroretinogramma che sarà anormale od estinto.
I recenti risultati degli studi di genetica molecolare sulla RP hanno dimostrato che esiste un comune meccanismo patogenetico, la degenerazione primitiva dei fotorecettori, che avviene sulla base di mutazioni di alcune delle proteine che costituiscono il ciclo della visione.
Epidemiologia
La RP è causa di disabilità visiva e cecità mondiale che affligge circa 1.5 milioni di pazienti con una prevalenza di circa 1:4000 che può variare con la localizzazione geografica.
Vengono differenziate le retiniti pigmentose in cui la malattia retinica è unica manifestazione, dalle retiniti pigmentose associate ad alterazioni di altri apparati, o sindromiche.
In passato venivano racchiuse sotto il nome di “retinite pigmentosa” anche una serie di distrofie retiniche che tuttavia, con il progredire delle conoscenze in ambito scientifico e diagnostico, si è scoperto non appartenere alla classica forma di RP, in quanto sono forme diverse che si manifestano alla nascita o nei primi mesi di vita (come la Leber congenital amaurosis, o LCA), le quali si caratterizzano per una degenerazione dei coni che precede quella dei bastoncelli, e per tale motivo vengono quindi definite distrofie cono-bastoncellari. Altra tipologia di distrofie retiniche differenti dalla RP sono le distrofie che interessano tipicamente la macula ed in fine disordini che non presentano un decorso di tipo progressivo come l’acromatopsia o la cecità notturna stazionaria (CSNB). Inoltre un 20–30% dei casi di RP si presenta come manifestazioni sindromiche che si associano ad alterazioni extra oculari. Tutte queste manifestazioni di RP formano un insieme di distrofie retiniche che si sovrappongono parzialmente sia clinicamente che geneticamente, per tale motivo in una prima classificazione furono incluse tutte sotto lo stesso tipo di patologia quale la RP.
Ereditarietà
Le forme genetiche di RP sono 4:
1. Eredità autosomica dominante
- può colpire maschi e femmine con pari frequenza
- non salta le generazioni
- La RP autosomica dominante rappresenta il gruppo delle forme meno gravi
2. Eredità autosomica recessiva
- la malattia colpisce con pari frequenza entrambi i sessi
- la malattia salta le generazioni. Questa ereditarietà risulta dalla forma recessiva ereditata da entrambi i genitori. Questa evenienza è molto rara stante la bassa frequenza del gene, però è spesso resa sensibilmente più probabile dalla consanguineità dei due genitori oppure anche dalla loro comune provenienza da un piccolo centro
3. Eredità legata al sesso: ereditarietà legata al cromosoma X
- risultano colpiti dalla malattia solo soggetti di sesso maschile che ereditano il gene patologico dalla madre, portatrice sana; data una donna in tale condizione, il rischio di malattia per ogni figlio maschio è del 50%.
- La RP legata al cromosoma X è tipicamente la forma più severa in termini di precocità di esordio, penetranza completa, progressione relativamente rapida, alta incidenza di miopia e di cataratta. In un piccolo numero di famiglie con RP tuttavia la malattia è relativamente mite nei pazienti maschi, probabilmente per ragioni di eterogeneità allelica o genetica.
4. Forme sporadiche di retinite pigmentosa: Sono circa il 30% di tutti i casi.
Classificazione
Si differenziano in base alle manifestazioni
- la RP primaria, in cui esiste solo l’interessamento oculare
- la RP che si ritrova associata a malattie extraoculari
La RP primaria, da un punto di vista fisiopatologico, viene suddivisa classicamente in due gruppi principali:
- rod-cone: le forme in cui la perdita di funzione dei bastoncelli precede quella dei coni
- cone-rod: le forme in cui la perdita di funzione dei coni precede quella dei bastoncelli
Tuttavia all’interno dell’ampio spettro delle distrofie retiniche ereditarie è preliminarmente importante classificare geneticamente le varie forme, per distinguere quelle appartenenti alla RP rispetto alle altre distrofie retiniche; che presumibilmente potrebbero avere un meccanismo patogenetico comune.
La classificazione genetica consente inoltre di fornire una prognosi al paziente definendo in prima approssimazione la gravità della forma di retinopatia, ed anche valutazione delle probabilità di trasmissione ai discendenti.
Infatti l’ampia eterogeneità tra i pazienti affetti da RP è dovuta all’elevato numero di difetti genetici associati alla RP. Nel 1990 è stato riportato il primo gene identificato coinvolto nella RP autosomica dominante: il gene della rodopsina (RHO), da allora fino al aggi sono note più di 80 mutazioni implicate nella RP non sindromica e sindromiche e ad ognuno di esso corrisponde a un sottotipo di RP gene-specifico che si manifesta con fenotipi di malattia differenti.
La maggioranza di questi geni codifica per proteine implicate nel ciclo della visione, e la mutazione genica più frequente, sia in termini di numero di mutazioni identificate (oltre 70), che di numero di pazienti colpiti (circa il 10%), riguarda il gene della rodopsina (il pigmento visivo dei bastoncelli) localizzato sul cromosoma 3 (3q21-q24), ed associato pressochè esclusivamente alla forma autosomica dominante (RP4).
Ognuno di questi geni codifica una proteina che svolge un ruolo nei processi vitali all’interno della neuroretina e/o dell’RPE (ad esempio, la cascata di fototrasduzione e la e del ciclo visivo) o in una struttura sottostante (per esempio, il cilium di connessione). Pertanto, una mutazione in un gene all’interno di una specifica via può causare la compromissione o l’interruzione dell’intera via o la sua totale interruzione. Per tale ragione varianti genetiche che modificano l’attività di una via possono aumentare l’eterogeneità clinica e/o genetica di malattie che coinvolgono una via con percorso comune.
Retinite pigmentosa non sindromica
La RP non sindromica ha una prevalenza maggiore rispetto a quella sindromica, ed è circa 1/4.000. Come già descritto è caratterizzata da una distrofia tipo bastoncelli-coni che insorge con cecità notturna, seguita dalla perdita progressiva della vista diurna, del campo visivo periferico, che può portare a cecità dopo diverse decadi. Di tutti i geni che causano la RP, ne sono stati identificati circa 50 geni/loci responsabili della RP non sindromica (forme autosomiche dominanti, autosomiche recessive, legate al cromosoma X e digeniche). La diagnosi clinica si basa sulla presenza di cecità notturna e sui difetti del campo visivo periferico, sulle lesioni nel fondo dell’occhio, sul tracciato elettroretinografico ipovoltato e sul progressivo peggioramento di questi segni. Quando si classifica una distrofia retinica è importante prendere in considerazione l’intero decorso della malattia, dato che alcuni fenotipi tendono a sovrapporsi nelle fasi finali., infatti la diagnosi molecolare per la Rp non sindromica non viene regolarmente eseguita a causa dell’estrema eterogeneità genetica della malattia. Le terapie in atto attualmente non sono in grado di arrestare la progressione della malattia o di restituire la vista; pertanto, la prognosi è infausta. L’approccio terapeutico permette di rallentare il processo degenerativo e consiste nella protezione dalla luce solare e nella terapia vitaminica, nel trattamento delle complicanze (cataratta e edema maculare) e nell’aiutare i pazienti a fare fronte ai problemi sociali e psicologici correlati alla cecità mediante la prescrizione di ausili per la lettura e lo svolgimento delle attività quotidiane al fine di rendere il paziente autosufficiente. Tuttavia, con i progressi in ambito scientifico stanno emergendo nuove strategie terapeutiche quali l’implemento mediante terapia genica, che al momento sono ancora in fase di studio, così come l’applicazione di protesi retiniche e l’ambito della neuroprotezione.
Retinite pigmentosa sindromica
Generalmente le mutazioni nei geni coinvolti nella funzione ciliare sono spesso coinvolti nelle forme sindromiche di RP.
Probabilmente, la ciliopatia più comune è la sindrome di Usher, che si presenta con un grado variabile di perdita dell’udito neurosensoriale.
Un’altra forma sindromica di RP ben nota è la sindrome di Bardet-Biedl; oltre alla retinopatia, i pazienti con questa sindrome possono presentare anche obesità, polidattilia postassiale, ipogonadismo, disfunzione renale e/o deterioramento cognitivo. Il tipo e l’entità di queste caratteristiche extra-oculari nel Bardet-Biedl possono variare notevolmente e dipendono per la maggior parte dal gene specifico coinvolto e dalla mutazione specifica all’interno di quel gene.
La RP sindromica è anche associata a disturbi metabolici e mitocondriali.
Le caratteristiche extra-oculari nella RP sindromica possono essere estremamente impercettibili (per esempio, un’alterazione dell’olfatto) o facilmente trascurate dal medico oculista (per esempio, nel caso di malattie cardiovascolari e/o renali. Pertanto, l’acquisizione di un’anamnesi attenta e approfondita che includa queste diverse anomalie extra-oculari è estremamente importante per ottenere una diagnosi. Tuttavia l’analisi genetica può rivelare mutazioni in un gene associato a forme sindromiche di RP, quando la valutazione clinica iniziale non indicava anomalie extra-oculari.
È bene tenere a mente che non tutte le anomalie extra-oculari sono correlate ad una malattia sindromica; queste anomalie devono corrispondere al gene coinvolto.
Un certo numero di geni associati alla RP non sindromica (ad esempio BBS1, CLRN1 e USH2A) possono causare anche RP sindromica.
Quadro clinico della retinite pigmentosa
La RP è caratterizzata dalla progressiva degenerazione dei fotorecettori e dell’epitelio pigmentato retinico (RPE) che portano alla cecità notturna, visione a tunnel e una graduale riduzione della visione centrale, con fenotipi che possono variare in base alla specifica mutazione genetica coinvolta. Un’accurata anamnesi familiare è molto importante in tutti i pazienti sospettati di RP. Tracciare un pedigree per ogni probando può essere molto utile al fine di valutare la modalità di ereditarietà e può anche avere conseguenze diagnostiche, oltre che essere da supporto per illustrare quali membri della famiglia sono a rischio di sviluppare la RP e/o indicare i soggetti per i quali si deve sospettare la non-penetranza.
Età di insorgenza e velocità di progressione
Nella presentazione “classica” della RP, la difficoltà di adattamento al buio inizia nell’adolescenza e la perdita visiva nel campo medio-periferico diventa evidente nella giovane età adulta. Tuttavia, l’età di insorgenza tra i pazienti affetti da RP varia notevolmente; pertanto, alcuni pazienti sviluppano una perdita visiva sintomatica nella prima infanzia, mentre altri possono rimanere relativamente asintomatica fino alla metà dell’età adulta. Inoltre spesso è difficoltoso determinare l’esatta età di insorgenza è poiché molti pazienti, soprattutto bambini, sono in grado di compensare la perdita visiva periferica.
In generale, i sottotipi di RP che si manifestano precocemente tendono a progredire più rapidamente. In generale, i pazienti con RP X-linked (5-15% dei pazienti con RP) hanno un decorso più grave rispetto ai pazienti con RP autosomica recessiva (50-60% dei pazienti con RP), mentre i pazienti con una forma autosomica dominante di RP (30-40% dei pazienti affetti da RP) hanno la migliore prognosi a lungo termine per quanto riguarda il mantenimento della visione centrale
Sintomi
I sintomi iniziali della RP comprendono cecità notturna (nyctalopia) e difficoltà di adattamento al buio. In alcuni casi, la RP può anche presentarsi con perdita del campo visivo medio-periferico, anche se questo è raramente segnalato come un sintomo precoce. Le funzioni della retina centrale rimangono relativamente conservate fino alle fasi finali della malattia, anche se le sue anomalie anatomiche possono comparire precocemente nel corso della malattia.
Nelle fasi finali della malattia, ed in genere quando il paziente raggiunge la mezza età, la degenerazione del cono centrale porta a un declino dell’acuità visiva. Tuttavia nonostante ciò la maggior parte dei pazienti con RP mantengono la capacità di percepire la luce grazie a una funzione maculare residua e/o alla o alla presenza di un’isola retinica periferica temporale conservata.
La fotopsia è un sintomo comune ma spesso trascurato che può essere molto fastidioso per i pazienti soprattutto negli stadi avanzati di malattia. Questo fenomeno può essere causato da una mancanza di impulsi nervosi afferenti in risposta alla degenerazione dei fotorecettori, o da un’attività spontanea di autosegnalazione come risultato del rimodellamento della retina interna.
I pazienti con RP possono anche manifestare fotofobia e discromatopsia.
Sia la miopia che l’ipermetropia elevate sono diffusi nei pazienti con RP
In alcuni sottotipi di RP possono essere presenti anomalie pigmentarie nella macula causate dall’atrofia dell’RPE centrale. Questi sottotipi sono sono associati a un declino più rapido dell’acuità visiva. Nei pazienti con i geni DHX38 o IDH3A l’atrofia maculare viene spesso definita pseudocoloboma maculare, da non confondere con un “vero” coloboma, che deriva da un difetto di chiusura della fessura embrionale durante lo sviluppo e spesso è accompagnato da altri difetti di chiusura.
Depositi di pigmenti maculari sono stati descritti in sono stati descritti nei sottotipi di RP ad esordio precoce dovuti a mutazioni in ABCA4 o RDH12. In questi sottotipi, i depositi di pigmento si estendono dalla periferia/metà periferia alla macula, differentemente dai sottotipi precedentemente descritti dove la macula rimane solitamente priva di pigmento.
Esame obiettivo
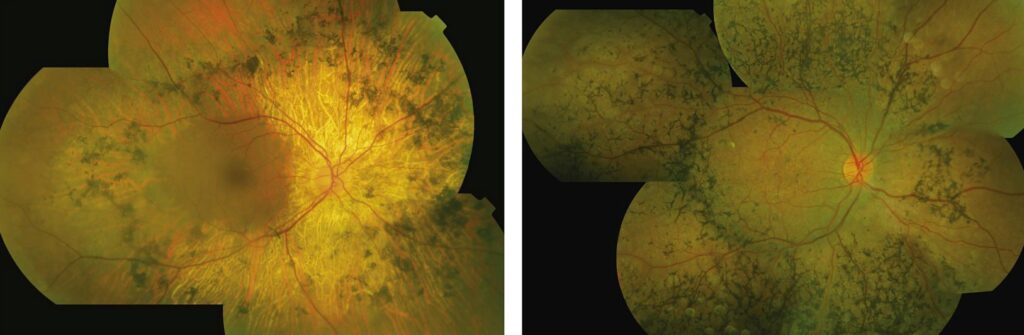
Le tre caratteristiche cliniche, segni distintivi della RP sono:
- la pigmentazione delle spicole ossee
- l’attenuazione dei vasi retinici
- il pallore cereo del nervo ottico.
Nelle fasi iniziali della RP l’esame del fundus può apparire normale, poiché queste alterazioni non sono ancora così marcate come i depositi di pigmento a forma di spicole ossee, o la minima attenuazione vascolare in presenza di un disco ottico ancora di aspetto normale.
Le caratteristiche spicole ossee possono avere un grado di iperpigmentazione variabile da un paziente all’altro e non riflette necessariamente la gravità della malattia. La loro pigmentazione è costituita da cellule RPE che si staccano dalla membrana di Bruch in seguito alla degenerazione dei fotorecettori e migrano verso siti perivascolari intraretinici. Queste spicole ossee spesso si formano nella media periferia, poiché in questa regione la concentrazione di cellule a bastoncello è più elevata (Fig.1).
Non si conosce con precisione cosa inneschi la migrazione degli RPE, si ipotizza che questa potrebbe essere innescata dalla riduzione della distanza tra i vasi retinici interni e l’RPE.
Tuttavia l’eziologia che sta alla base dell’attenuazione dei vasi retinici nella RP rimane non è chiara. Vi sono diverse ipotesi:
a) la prima ipotesi riguarda la ridotta richiesta metabolica la quale si verifica in seguito alla degenerazione delle cellule ganglionari, che a loro volta è secondaria alla perdita dei fotorecettori.
b) la seconda ipotesi attribuisce la perdita di fotorecettori che consumano ossigeno a uno stato iperossico della retina interna rimanente, che porta alla vasocostrizione e alla riduzione del flusso sanguigno nei vasi retinici.
Infatti la morte delle cellule dei fotorecettori comporta una perdita di input sinaptico e la conseguente diminuzione dei fattori trofici, la quale provoca una riduzione del metabolismo degli strati interni della retina. Questo fenomeno comporta il rimodellamento vascolare e la conseguente attenuazione dei vasi.
Il disco ottico sviluppa tipicamente un pallore cereo con il progredire della malattia, a causa dalla formazione di cellule gliali sia sulla superficie che all’interno del disco ottico, con conseguente aumento della riflessione della luce.
Esami strumentali
Perimetria
La perdita progressiva del campo visivo è una caratteristica della RP. Questa si manifesta bilaterale in modo simmetrico ed inizia tipicamente con scotomi isolati nelle aree medio-periferiche, che gradualmente si uniscono a formare uno scotoma anulare parziale o completo. Con il progredire della malattia, questo scotoma anulare si estende sia verso l’esterno che verso l’interno. La perimetria cinetica è la più adatta per la valutazione della perdita di campo visivo periferico.
Visione dei colori
Inizialmente la visione dei colori può essere normale. Nelle fasi avanzate della malattia possono essere presenti le discromatopsie, in particolare i difetti di visione dei colori blu-giallo (in cui i pazienti presentano difficoltà a distinguere le tonalità del blu dal verde e del giallo-verde dal viola). La disfunzione dei coni blu è stata attribuita alla scarsità di questi coni a breve lunghezza d’onda sulla fovea; per tale motivo nella RP i difetti acquisiti della visione dei colori di tipo III (blu) sono più diffusi rispetto a quelli di tipo I (rosso-verde). A causa di questa distribuzione non uniforme, la perdita della funzione retinica pericentrale può portare alla tritanopia (cecità ai colori giallo-blu).
La perdita dell’acuità visiva, associata alla degenerazione dei fotorecettori centrali aumenta la probabilità di sviluppare un difetto cromatico di tipo I.
Adattometria al buio
Un’anomalia della soglia di adattamento al buio è un tratto distintivo della RP. La soglia dei bastoncelli è spesso aumentata a causa di una diminuzione della sensibilità di questi fotorecettori che si associa ad un recupero prolungato della sensibilità stessa.
Ricordiamo che in questa patologia sono caratteristiche
- un aumento della soglia dei coni e dei bastoncelli
- un ritardo nel raggiungimento della soglia asintotica dei bastoncelli
- la perdita completa della funzione dei fotorecettori a bastoncello
Elettroretinografia
I test elettrofisiologici a tutto campo – secondo le linee guida ISCEV (http://www.iscev.org/standards) – aiutano la diagnosi e sono essenziali per la valutazione quantitativa della gravità della malattia, nonché per il suo monitoraggio. Le anomalie dell’elettroretinogramma (ERG) si manifestano precocemente e precedono i sintomi della cecità notturna e le anomalie del fondo. Su l’ERG adattato al buio, con flash luminoso (combinato bastoncello-cono), l’onda a è subnormale. Inoltre, le risposte isolate dei bastoncelli a un flash luminoso (scotopico) sono ritardate, ridotte o assenti in una registrazione ERG a tutto campo. Anche le risposte dei coni possono essere influenzate nelle prime fasi della RP, ma questo fenomeno è tipicamente ritardato rispetto all’insorgenza della disfunzione dei bastoncelli. Quando è presente, la disfunzione del cono si manifesta nell’ERG adattato alla luce (fotopico) con una risposta ritardata e ridotta a un flash luminoso e a stimoli flicker a 30 Hz. Anche i potenziali oscillatori possono essere ridotti nei pazienti con RP.
Con il progredire della malattia, l’ERG a tutto campo può diventare non registrabile nonostante un campo visivo residuo. In queste circostanze, una soglia di stimolo a tutto campo (FST), un test veloce che non richiede la fissazione del paziente, o un ERG multifocale (mfERG) possono essere ancora in grado di suscitare risposte e possono quindi essere utilizzati per seguire la progressione della malattia.
Complicanze associate alla retinite pigmentosa
Diverse altre condizioni oculari, alcune delle quali trattabili, sono spesso associate alla RP. Per esempio, i pazienti con RP ad esordio precoce possono presentare anche nistagmo e l’errore di rifrazione associato alla malattia è altrettanto comune.
Le complicazioni maculari possono includere:
- edema maculare cistoide (ECM) 50% dei pazienti con RP
- foro maculare
- formazione di membrana epiretinica 36% dei pazienti con RP
Sono stati proposti diversi meccanismi che possono contribuire alla formazione di ECM:
- la rottura della barriera emato-retinica
- l’alterazione della funzione del meccanismo di pompaggio dell’RPE
- edema e disfunzione delle cellule di Müller
- anticorpi antiretinici
- trazione vitreomaculare
L’edema maculare cistoide è un reperto comune tra i pazienti affetti da RP ed è prevalente in tutte le fasce. La prevalenza riportata tra i pazienti con una forma autosomica dominante di RP è relativamente alta. Un rapporto ha anche suggerito un’associazione tra ECM e pazienti di sesso femminile. Una combinazione di ECM e cellule nel corpo vitreo è stata segnalata in bambini con alcune varianti genetiche, dove la combinazione di reperti clinici può portare a una diagnosi errata di uveite intermedia, in particolare nei bambini affetti da RP in cui le anomalie retiniche sono sottili o addirittura assenti.
I meccanismi che comportano la formazione della membrana epiretinica sono:
- una proliferazione idiopatica delle cellule gliali preretiniche
- potrebbe essere secondaria ad un processo infiammatorio
infatti la componente infiammatoria nella RP non è nuova, come testimonia la parola “retinite” nel nome, e si ritiene generalmente che sia secondaria proprio alla morte delle cellule dei fotorecettori, principale meccanismo patologico di questa distrofia.
La cataratta sottocapsulare posteriore è un’altra manifestazione che può influire significativamente sulla visione e si verifica in circa il 45% dei pazienti affetti da RP, il meccanismo alla base della sua formazione è attualmente sconosciuto.
Un’altra anomalia del vitreo che può verificarsi nella RP è la presenza di cisti vitreali (6% dei pazienti con RP).
Inoltre, drusen della testa del nervo ottico e/o drusen dello strato di fibre del nervo ottico sono state segnalate nel 9% dei pazienti con RP.
Imaging retinico
Tomografia a coerenza ottica
I cambiamenti nella morfologia retinica determinati da questo tipo di distrofia si riflettono in una tomografia a coerenza ottica a dominio spettrale (SD-OCT), esame che permette di analizzare gli strati retinici mediante un fascio di luce laser.
Il cambiamento più precoce che si riscontra è l’accorciamento dei segmenti esterni dei fotorecettori, che si riflette nella disorganizzazione degli strati retinici esterni.
Con il progredire della RP l’assottigliamento dei segmenti esterni è accompagnato da una diminuzione dello spessore dello strato nucleare esterno, che è lo strato retinico che contiene i nuclei dei fotorecettori. Gli ultimi stadi della RP sono caratterizzati dalla perdita completa dello strato esterni della retina (Fig.2).
Gli strati retinici interni, tra cui lo strato delle cellule ganglionari, rimangono relativamente ben conservati e con il progredire della malattia questo strato potrebbe andare incontro a rimodellamento neuronale-gliale della retina, in risposta all’assottigliamento della retina esterna. Quindi questo processo potrebbe comportare un ispessimento degli strati retinici interni
Altro reperto comune è la presenza di Focolai iper-riflettenti negli strati retinici esterni che rappresentano le cellule dell’epitelio pigmentato, le quali dopo essere andate incontro a degenerazione, hanno migrato verso gli strati più superficiali.
L’imaging OCT può anche essere prezioso per diagnosticare altre anomalie maculari presenti in fino alla metà dei pazienti affetti da RP, come appunto l’edema maculare cistoide (ECM) che è il reperto più comune, seguito da formazione di membrana epiretinica, sindrome da trazione vitreomaculare e foro maculare.
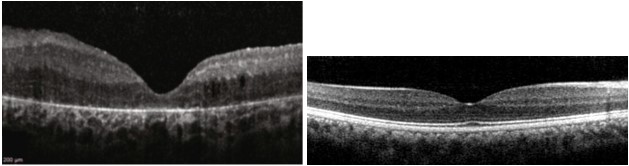
retinici esterni: strato dei fotorecettori ed RPE assente (sinistra). Confronto con scansione OCT in
un paziente sano (destra).
Imaging in autofluorescenza del fondo
L’autofluorescenza del fondo (FAF) può rivelare un’alterazione del metabolismo dell’RPE altrimenti non rilevabile. Esistono due metodiche la SW FAF (lunghezza d’onda corta) che utilizza luce blu o verde per individuare il segnale proviene principalmente dalle molecole di lipofuscina, che è il materiale di scarto delle cellule presenti nell’RPE. Invece la NIR FAF che utilizza lo spettro dell’infrarosso, visualizza il segnale di autofluorescenza che proviene dalla melanina dell’RPE.
La FAF è sempre più sempre più utilizzata per valutare e monitorare la progressione della RP. Il reperto caratteristico presente nel 50-60% dei pazienti che si ottiene con questo esame è un anello foveale anormale di autofluorescenza aumentata non visibile all’oftalmoscopia. Questo rappresenta una zona di transizione tra la funzione retinica anormale e quella normale. La funzione è relativamente normale all’interno dell’anello e assente all’esterno dell’anello. Questo reperto è visibile sia con la SW FAF che con la NIR FAF poiché con entrambe le metodiche è possibile visualizzare il materiale delle cellule dell’RPE che sono degenerate in questa malattia (Fig. 3).
Negli stadi più avanzati di malattia l’anello può disperdersi, e questo fenomeno è correlato a una perdita diffusa di sensibilità e dell’acuità visiva.
La microperimetria nei pazienti con RP mostra che la sensibilità visiva è relativamente conservata all’interno dell’anello, ridotta nella zona dell’anello stesso, e diminuita o non registrabile nella regione esterna all’anello.
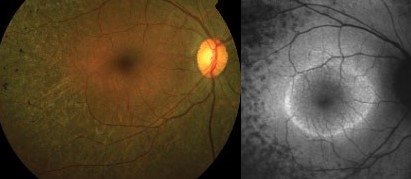
con spicole ossee, a destra si noti l’anello parafoveale di FAF aumentato all’intero dell’anello e
FAF diminuito al di fuori.
Biochimica della fototrasduzione
La fototrasduzione comprende una cascata di reazioni che vengono innescate in seguito all’eccitazione della molecola dell’opsina da parte di un fotone. Questo evento permette la trasmissione del segnale elettrico attraverso il nervo ottico fino alla corteccia visiva. La cascata di reazioni che avvengono nei due tipi di fotorecettori – coni e bastoncelli – è in gran parte simile. Le differenze che si riscontrano tra i due sono dovute alle diverse sensibilità che hanno rispettivamente i coni (per condizioni di luce intensa) rispetto ai bastoncelli (per condizioni di luce crepuscolare).
Bastoncelli: in questo tipo di fotorecettori la rodopsina è costituita dall’apoproteina opsina e dal cromoforo 11-cis-retinale. L’interazione con il fotone determina la conversione dell’11-cis-retinale nell’isomero tutto-trans-retinale. Questo cambiamento conformazionale determina un cambiamento nella struttura della rodopsina in quella della metarodopsina fotoattiva. La metarodopsina II attiva la proteina G transducina che attiva la guanosina monofosfato ciclica (cGMP) fosfodiesterasi che idrolizza il cGMP per formare il 5′-GMP. Questo processo diminuisce la concentrazione di cGMP nel citoplasma del fotorecettore, determinando la chiusura dei canali cationici a rilascio di cGMP situati nella membrana plasmatica. La chiusura dei canali determina l’iperpolarizzazione della membrana plasmatica proprio a causa di una forte diminuzione della concentrazione di calcio intracellulare. L’iperpolarizzazione della membrana plasmatica porta a una diminuzione del rilascio di glutammato alla sinapsi del fotorecettore. Il rilascio di glutammato è inibito in condizioni di luce.
Dopo la fototrasduzione, il sistema ritorna alla fase di pre-fotoattivazione. attraverso le seguenti fasi:
1- fosforilazione della metarodopsina II da parte della rodopsina chinasi;
2- la dissociazione dell’all-trans-retinale dal pigmento visivo e la conversione in 11-cis-retinale attraverso il ciclo visivo (retinoide)
3- il ritorno del cGMP intracellulare a livelli normali che viene attivato dalla guanilato ciclasi.
Dopo che tutto il trans-retinico si è dissociato dall’opsina, l’11-cis-retinico si lega all’opsina per produrre la rodopsina. La rodopsina viene quindi fosforilata dalla proteina fosfatasi 2A. Pertanto, al buio a livello dei bastoncelli, la rodopsina è prevalentemente allo stato non fosforilato.
Coni: Esistono due differenze principali tra bastoncelli e coni per quanto riguarda la fototrasduzione
1- le cellule a cono esprimono tre diverse opsine, ognuna delle quali è specifica – anche se meno sensibile per una determinata lunghezza d’onda.
2- le opsine nelle cellule del cono hanno una cinetica più veloce di quella dei bastoncelli, questo si traduce in una fase di recupero più breve.
Il ciclo visivo
Il ciclo visivo è un processo complesso che si concentra sulla rigenerazione 11-cis-retinale dall’all-trans-retinale prodotto nella cascata di fototrasduzione. Tale rigenerazione si verifica contemporaneamente alla trasduzione del segnale stesso.
La molecola 11-cis-retinale è un derivato della vitamina A (all-trans-retinolo). Quest’ultima viene assorbita si accumula nell’EPR e viene convertita in 11-cis-retinale.
Al momento della fotoattivazione, al livello dei fotorecettori, la molecola tutto-trans-retinale viene rilasciata dal pigmento visivo (il quale viene attivato nel lume dei dischi del segmento esterno delle fotorecettori). Successivamente, a livello del citoplasma del fotorecettore, il tutto-trans-retinale viene ridotto a tutto-trans-retinolo dall’enzima all-trans-retinal deidrogenasi. Il tutto-trans-retinolo viene quindi rilasciato nello spazio sottoretinico, dove si lega alla proteina legante i retinoidi (IRBP) e viene trasportato all’EPR. Nel citoplasma della cellula EPR, l’all-trans-retinolo si lega proteina legante il retinolo cellulare e viene re-isomerizzato ad 11-cis-retinale. L’11-cis-retinale risultante viene poi trasportato nel citoplasma del fotorecettore da IRBP. Una volta rientrato nel fotorecettore, l’11-cis-retinale si lega all’opsina per formare una nuova molecola di rodopsina.
Questo percorso, noto come ciclo visivo canonico, catalizza la re-isomerizzazione della retina nelle cellule dei bastoncelli.
Trasporto ciliare
Le cilia sono sottili proiezioni longitudinali a base di microtubuli che si estendono dalla superficie cellulare. A livello oculare le cellule dei fotorecettori contengono un cilium sensoriale altamente specializzato, che rappresenta la struttura altamente specializzata in cui avviene la fototrasduzione. Il cilium di connessione permette la comunicazione tra il segmento esterno ed interno del fotorecettore, mediante il processo di trasporto intraflagellare (IFT), un sistema di trasporto bidirezionale, guidato dalle proteine motrici chinesine, che sfrutta i microtubuli per lo spostamento dei componenti biosintetiti del ciclo visivo.
Molti geni associati alla RP non sindromica codificano proteine che sono coinvolte in vari aspetti del trasporto ciliare. Sono state identificate mutazioni in circa 30 geni codificanti per proteine ciliari essenziali per la funzione/regolazione dell’IFT o nella struttura ciliare. Ad esempio il complesso BBSome che causa la sindrome di Bardet-Biedl (BBS), o il gene RPGR che è responsabile del 70-90% dei casi di RP X-linked e del 10-20% di tutti i casi di RP.
Struttura del segmento esterno
Nella struttura del fotorecettore retinico si riconosce un segmento esterno contenente un compartimento altamente specializzato costituito da pile di dischi intracellulari (nelle cellule a bastoncello) o di lamelle (nelle cellule a cono), strutture che concorrono anch’esse al ciclo visivo.
Alcuni sottotipi di RP non sindromica sono associati a mutazioni delle proteine che sono coinvolte nello sviluppo e/o nell’orientamento dei dischi del segmento esterno.
La matrice interfotorecettoriale
La matrice interfotorecettoriale riempie lo spazio sottoretinico dove si trovano i segmenti interni ed esterni delle cellule fotorecettrici. La matrice svolge un ruolo importante nei processi di regolazione del trasporto di ossigeno e nutrienti ai fotorecettori e nel metabolismo della retina (retinoidi).
La matrice interfotorecettoriale è composta da proteine e carboidrati che vengono secreti dai fotorecettori e dalle cellule RPE. Tra i suoi componenti principali si trova l’acido ialuronico che forma una rete tridimensionale di connessione per le cellule tra cui quelle dell’RPE tramite proteine contenenti RHAMAM (recettore per la motilità).
Inoltre, la matrice extracellulare potrebbe avere ruolo chiave nei processi clinici patologici della RP. Infatti 3 geni (IMPG2, RBP3 e EYS) associati alla RP non sindromica codificano proteine che si legano alla rete dell’acido ialuronico.
1. Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018 Sep;66:157-186. doi: 10.1016/j.preteyeres.2018.03.005. Epub 2018 Mar 27. PMID: 29597005.
2. Isabelle Audo, MD, A. Robson, P. Hykin, A. Bird. FAF Imaging for Retinal Diseases, Review of ophthalmology, august 2005.
3. Orphanet, Rare diseases https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=659
