In questo articolo sul retinoblastoma, imparerai:
- La genetica del retinoblastoma
- Come diagnosticare precocemente il retinoblastoma
- Come distinguerlo da altre patologie oculari dell’età pediatrica
- Come trattare il retinoblastoma
Introduzione
Il retinoblastoma è la più comune neoplasia maligna intraoculare dell’infanzia, tuttavia, rispetto ad altri tumori dell’età pediatrica, esso è da considerarsi un tumore raro, rappresentando solo il 3-4% di tutte le neoplasie infantili. Il retinoblastoma è un tumore maligno che origina a partire dalle cellule staminali retiniche primitive o dai precursori dei fotorecettori retinici.
L’incidenza globale si attesta intorno a 1 caso su 16.000-18.000 nati vivi (0,01-0,04%). Non si è individuata una predisposizione di genere, essendo l’incidenza sovrapponibile nel sesso maschile e in quello femminile.
L’età di insorgenza di solito varia dalla nascita ai primi 3 anni di vita, con un’età media alla diagnosi che risulta essere più precoce (7-12 mesi) nei casi bilaterali e più tardiva (18-24 mesi) nei casi unilaterali.
Genetica
Dal punto di vista genetico, il retinoblastoma costituisce un ottimo esempio per comprendere la genetica del cancro. La tumorigenesi è un processo che prevede una serie di mutazioni genetiche in sequenza e solo alcune di queste sono davvero rilevanti perché si abbia una trasformazione maligna.
Studiando il pattern di ereditarietà del retinoblastoma, Knudson per primo ha proposto il “two-hit” model o “ipotesi dei due colpi”, con cui si prefiggeva di spiegare come un allele mutato di un gene oncosoppressore, ereditato come tratto dominante a cui, in seguito, si aggiungesse una seconda inattivazione dell’allele sano, potesse indurre lo sviluppo del retinoblastoma.
L’ipotesi avanzata da Knudson è stata successivamente confermata dal team guidato da Weinberg e Dryja che, nel 1986, hanno identificato e clonato il gene RB1. Come previsto da Knudson, una copia del gene RB1(190 Kb), localizzato sul cromosoma 13q14, risulta mutato nelle cellule della linea germinale di soggetti predisposti, mentre entrambe le copie del gene risultano mutate nei soggetti che presentano il retinoblastoma.
L’“ipotesi dei due colpi” prevede che siano necessari due eventi mutageni affinché si sviluppi il tumore: nelle forme ereditarie, i pazienti presentano da subito una mutazione nella linea germinale di RB1 (primo colpo) a cui fa seguito una seconda mutazione (secondo colpo), mentre nelle forme non ereditarie, i pazienti acquisiscono in maniera sporadica due diverse mutazioni a carico di RB1 nelle cellule somatiche della retina in via di sviluppo.
Ne consegue, pertanto, che il retinoblastoma si può presentare sotto forma di tre diverse entità:
- retinoblastoma familiare: il 10% dei bambini che presentano il retinoblastoma eredita una copia mutata del gene RB1 (primo colpo) da un genitore, di conseguenza ogni cellula di questi individui presenta un allele mutato. La mutazione a carico del secondo allele di RB1 (secondo colpo) si verifica nelle cellule retiniche dopo il concepimento.
- retinoblastoma sporadico ereditabile: il 30% dei bambini affetto da retinoblastoma presenta in ogni sua cellula corporea un allele RB1 mutato. La differenza rispetto alla situazione precedente è che questi bambini non ereditano l’allele mutato da uno dei due genitori, ma la mutazione dell’allele è una mutazione acquisita insorta ex novo a carico delle cellule germinali. Pertanto, anche se questi bambini non ereditano la copia mutata da un genitore, essi stessi potranno tuttavia trasmetterla ai loro figli in futuro.
- retinoblastoma non ereditario: il 60% dei pazienti presenta una forma di retinoblastoma non ereditario, ossia il tumore si sviluppa come risultato di mutazioni che avvengono a carico di entrambi gli alleli di RB1 e che si verificano in una singola cellula retinica dopo il concepimento e tali mutazioni determinano una perdita di funzione della proteina codificata dal gene RB1.
Il gene RB1, costituito da 27 esoni, codifica per una proteina di 928 aminoacidi nota come pRB che gioca un ruolo fondamentale come regolatore del ciclo cellulare: essa, infatti, legando i fattori di trascrizione della famiglia E2F, va ad inibire l’attivazione dei geni che inducono la proliferazione cellulare e, di conseguenza, impedisce la transizione della cellula dalla fase S alla fase G1 del ciclo cellulare.
Nel retinoblastoma, la mutazione a carico del gene RB1 determina la disfunzione o l’assenza della proteina pRB e ciò comporta una costante attivazione dei fattori di trascrizione E2F che, di conseguenza, inducono la cellula a proliferare continuamente, causando la formazione del tumore.
Ad oggi sono state individuate numerose mutazioni genetiche a carico di RB1, quali riarrangiamenti cromosomici, delezioni di esoni, fenomeni di ipermetilazione della regione del promotore genico, mutazioni a singolo nucleotide e sostituzioni.
Le mutazioni più frequenti della linea germinale sono mutazioni no-sense o frame-shift all’interno degli esoni 2-25 che portano o alla mancata formazione di pRB o alla perdita di funzione della proteina.
Clinica
Il più comune segno clinico di presentazione del retinoblastoma è la leucocoria (riflesso pupillare bianco) nel 60% dei casi, seguito da strabismo nel 20% dei casi. Il retinoblastoma origina dalla retina in via di sviluppo e tende a presentarsi sotto forma di una massa bianca sollevata e cupoliforme a cui spesso possono associarsi coinvolgimento vitreale e/o sottoretinico.
Quando il tumore raggiunge dimensioni tali da superare la quantità di ossigeno necessaria alla sopravvivenza delle cellule tumorali, hanno inizio processi di necrosi retinica con comparsa di aree di calcificazione intratumorali.
Il retinoblastoma può avere una crescita di tipo
- endofitico: il tumore cresce e aggetta nel gel vitreale. Di solito questi tumori hanno un colorito biancastro senza eccessiva vascolarizzazione.
- esofitico: il tumore cresce al di sotto della retina andando a invadere la coroide e potendo determinare un distacco di retina essudativo.
- misto: sia endofitico, sia esofitico.
infiltrante: il tumore non si sviluppa in profondità, ma piuttosto va ad infiltrare la retina, espandendosi in larghezza. E’ un pattern di crescita raro che si osserva quando il tumore viene diagnosticato in età più tardiva. Può essere difficile distinguerlo da alcune forme di endoftalmite e/o uveite.
Diagnosi
- anamnesi: nei pazienti che arrivano alla visita oculistica per sospetto di retinoblastoma, è importante un’accurata anamnesi che deve tener conto di aspetti quali l’età gestazionale e il peso alla nascita, il tipo di parto, infezioni o farmaci durante la gravidanza. Si procede indagando lo stato di salute attuale del bambino, prendendo in considerazione anche l’ambiente in cui vive (presenza di eventuali animali domestici), valutando lo stato di salute di eventuali fratelli e storia di patologie oculari nella famiglia.
- segmento anteriore: Occorre valutare l’atteggiamento del bambino al momento della visita, considerando eventuali anormalità cranio-facciali, la presenza di leucocoria, strabismo, edema periorbitario. La valutazione dell’acuità visita dipenderà dall’età del bambino e dal suo grado di collaborazione. E’ importante esaminare i riflessi pupillari, la presenza di eterocromia dell’iride e la valutazione del riflesso rosso della retina. Spesso i genitori si accorgono dell’assenza del riflesso rosso esaminando delle fotografie.
- segmento posteriore (in midriasi): l’esame del polo posteriore è più facilmente eseguibile con l’oftalmoscopio indiretto. A seconda dell’età, il bambino può collaborare o può essere necessario l’aiuto dei genitori. L’esame a questo punto permette o di confermare il sospetto di retinoblastoma o, nel caso non si riesca a effettuare un’adeguata valutazione del fondo oculare, si procederà all’esame del segmento posteriore in anestesia generale.
- ecografia oculare: l’ecografia può essere eseguita in modalità A o B scan per visualizzare la presenza di una massa, di una calcificazione o di un distacco retinico associato. Spesso l’ecografia oculare viene svolta sotto anestesia in modo da valutare lo stato dell’orbita e misurare lo spessore della neoformazione.
- esami ancillari:
- fotografia del segmento anteriore/retinografia: utili ai fini di documentare lo stato del segmento anteriore e posteriore e valutare eventuali modifiche nel tempo.
- angiografia con fluoresceina sodica (FA): utile durante l’anestesia generale nei casi dubbi al fine di poter effettuare una corretta diagnosi differenziale. In presenza di un retinoblastoma, alla fluorangiografia si potranno apprezzare vasi tumorali dilatati che attraversano una massa talvolta ipo o iper fluorescente con leakage di colorante in relazione alle dimensioni della lesione. Questo esame è dirimente nella diagnosi differenziale con la malattia di Coats in quanto, in questo secondo caso, si osservano vasi retinici teleangectasici che, a differenza del retinoblastoma, rimangono sul piano retinico e risultano circondati da ampie aree di retina ischemica.
- elettroretinogramma (ERG): viene usato al fine di monitorare la funzione retinica prima, durante e dopo la chemioterapia intra-arteriosa.
- tomografia computerizzata (TC) dell’orbita: le scansioni tomografiche storicamente sono state molto utili per identificare le aree di calcificazione presenti nel contesto del retinoblastoma. Tuttavia, attualmente, si cerca di evitare di effettuare la TC al fine di non esporre i bambini ad alte dosi di radiazioni ionizzanti.
- risonanza magnetica (MRI) di encefalo e orbite con e senza contrasto: rappresenta attualmente il gold standard per la diagnosi.
Diagnosi differenziale
- amartoma astrocitico: i pazienti presentano una neoformazione di colore grigio-giallo traslucida che coinvolge il polo posteriore, spesso in prossimità del nervo ottico. Sono lesioni sessili di piccole dimensioni che spesso contengono aree di calcificazione al momento della diagnosi. Spesso presentano una fine vascolarizzazione superficiale che viene facilmente evidenziata con l’angiografia con fluoresceina.
- vascolarizzazione fetale persistente (PFV): in condizioni fisiologiche, la vascolarizzazione fetale che nutre il cristallino e la retina in via di sviluppo tende a regredire una volta che si sono correttamente sviluppati i vasi retinici. Qualora tale regressione non si verifichi, si configura il quadro di “vascolarizzazione fetale persistente” che è responsabile di circa il 5% dei casi di cecità infantile nei paesi occidentali. Può essere anteriore o posteriore. Anteriore se è confinata al segmento anteriore e spesso coinvolge il cristallino. Si osserva una massa retrolentale costituita dai processi ciliari allungati che si evidenzia come leucocoria e spesso è complicata da cataratta e glaucoma. Posteriore se è limitata al segmento posteriore e il cristallino di solito è trasparente. Si può presentare con leucocoria, strabismo e, occasionalmente, con nistagmo. Si osserva una massa di vitreo e retina che si estende dall’ora serrata al disco ottico.
- malattia di Coats: si tratta di una telangectasia retinica idiopatica caratterizzata da essudazione intra e/o sottoretinica. DI solito è unilaterale e colpisce preferenzialmente il sesso maschile nella prima – seconda decade di vita, pertanto tende a presentarsi molto più tardivamente del retinoblastoma. La leucocoria causata dalla malattia di Coats è spesso più giallastra che nel retinoblastoma a causa della presenza dell’essudazione sottoretinica e infatti si tende a parlare di “xantocoria” (riflesso pupillare giallo) più che di leucocoria.
- toxocariasi: frequentemente viene scambiata per retinoblastoma. Si può presentare in tre forme: 1) granuloma maculare 2) granuloma periferico 3) endoftalmite. Un segno tipico della toxocariasi è la presenza di trazione retinica e/o vitreale.
- retinopatia del prematuro (ROP): nei casi avanzati di retinopatia del prematuro, l’estesa proliferazione fibrovascolare può causare leucocoria, ponendo così il sospetto di retinoblastoma. La diagnosi di ROP è facilitata dalla storia di prematurità e basso peso alla nascita.
- sindromi retiniche ereditarie:
- vitreopatia essudativa familiare (FEVR): rara malattia genetica dovuta ad un’alterata formazione della retina durante lo sviluppo embrionale che si caratterizza per la presenza di avascolarità della retina temporale associata a proliferazione fibrovascolare periferica con coinvolgimento di solito bilaterale.
- malattia di Norrie: è dovuta alla mutazione del gene NDP localizzato sul cromosoma X (XP11.4). I pazienti presentano displasia retinica bilaterale con distacco di retina trazionale, emorragia endovitreale e presenza di una massa retrolentale di solito costituita da retina staccata (pseudoglioma). Si può associare a cataratta e microftalmia. I bambini affetti possono presentare anche ritardo mentale e sordità congenita. L’ereditarietà è X-linked recessiva pertanto solo i maschi ne sono affetti.
- incontinentia pigmenti: è una rara malattia genetica X-linked dominante che colpisce la pelle, i denti, le ossa, gli occhi e il sistema nervoso centrale. A livello oculare si riscontra una proliferazione fibrovascolare che risulta in distacco di retina trazionale associato a presenza di microaneurismi e neovascolarizzazione retinica.
- coloboma corioretinico: si tratta di una condizione congenita dovuta alla mancata chiusura della fossetta ottica durante lo sviluppo embrionale a cui consegue un’assenza focale di retina e coroide. La posizione del coloboma è in genere inferiore. La sclera esposta appare in genere biancastra a margini pigmentati.
Stadiazione
Un sistema di stadiazione dei tumori condiviso è essenziale per effettuare una corretta diagnosi e, di conseguenza, per pianificare il trattamento più adeguato e per stabilire la prognosi.
Nella stadiazione della maggior parte dei tumori, una biopsia tissutale e/o il dosaggio di eventuali marcatori tumorali giocano un ruolo fondamentale.
Nel panorama dei tumori dell’età pediatrica, il retinoblastoma rappresenta un tumore unico per una serie di aspetti:
- Il trattamento si prefigge due obiettivi: salvare il globo oculare e la vita del paziente.
- La biopsia è controindicata per il rischio di disseminazione tumorale.
- La gestione del retinoblastoma è di pertinenza oculistica, pertanto l’oncologo e il pediatra non possono prescindere dalla valutazione oculistica per procedere con la diagnosi e il corretto trattamento della neoplasia.
In considerazione delle suddette caratteristiche del retinoblastoma, non è mai stato possibile adottare per questo tumore un sistema di stadiazione simile a quelli in uso per le altre neoplasie solide dell’infanzia.
Il primo sistema di classificazione del retinoblastoma risale al 1960 e prende il nome di Classificazione di Reese-Ellsworth. Tale sistema è stato utilizzato per decenni: esso prevedeva una classificazione del retinoblastoma in gruppi sulla base della probabilità di effettuare un trattamento conservativo (e quindi di poterlo trattare con la radioterapia che, al tempo, rappresentava la prima opzione terapeutica).
Stadiazione
Un sistema di stadiazione dei tumori condiviso è essenziale per effettuare una corretta diagnosi e, di conseguenza, per pianificare il trattamento più adeguato e per stabilire la prognosi.
Nella stadiazione della maggior parte dei tumori, una biopsia tissutale e/o il dosaggio di eventuali marcatori tumorali giocano un ruolo fondamentale.
Nel panorama dei tumori dell’età pediatrica, il retinoblastoma rappresenta un tumore unico per una serie di aspetti:
- Il trattamento si prefigge due obiettivi: salvare il globo oculare e la vita del paziente.
- La biopsia è controindicata per il rischio di disseminazione tumorale.
- La gestione del retinoblastoma è di pertinenza oculistica, pertanto l’oncologo e il pediatra non possono prescindere dalla valutazione oculistica per procedere con la diagnosi e il corretto trattamento della neoplasia.
In considerazione delle suddette caratteristiche del retinoblastoma, non è mai stato possibile adottare per questo tumore un sistema di stadiazione simile a quelli in uso per le altre neoplasie solide dell’infanzia.
Il primo sistema di classificazione del retinoblastoma risale al 1960 e prende il nome di Classificazione di Reese-Ellsworth. Tale sistema è stato utilizzato per decenni: esso prevedeva una classificazione del retinoblastoma in gruppi sulla base della probabilità di effettuare un trattamento conservativo (e quindi di poterlo trattare con la radioterapia che, al tempo, rappresentava la prima opzione terapeutica).
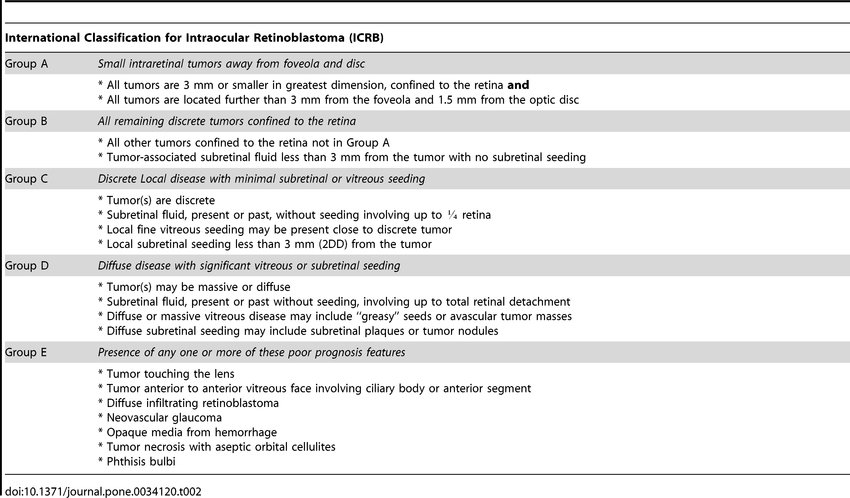
Nei primi anni Novanta, la radioterapia è stata a poco a poco soppiantata dalla chemioterapia endovenosa e, pertanto, il sistema di classificazione di Reese-Ellsworth è stato sostituito con una classificazione che rispecchiasse maggiormente la nuova pratica clinica.
Nel 2005, nell’era della chemioterapia sistemica quale trattamento di prima scelta per il retinoblastoma, è stata pubblicata l’International Intraocular Retinoblastoma Classification (IIRC).
Shields e colleghi hanno in seguito sviluppato una classificazione modificata, nota come International Classification of Retinoblastoma (ICRB), che si distingue dalla precedente nella definizione degli stadi avanzati di retinoblastoma.

L’American Joint Committee on Cancer (AJCC) ha pubblicato, a propria volta, un sistema di stadiazione universale valido per tutte le neoplasie, incluso il retinoblastoma, sulla base dei criteri TNM. L’ottava edizione è attualmente la più recente e la più utilizzata.
Trattamento
I tassi di sopravvivenza per i pazienti con retinoblastoma sono aumentati drasticamente nel secolo scorso, con una sopravvivenza a 5 anni che raggiunge il 95-99% nei paesi sviluppati. Questi risultati sono stati possibili grazie ai significativi progressi che si sono fatti nell’ambito della gestione del retinoblastoma.
Chemioterapia:
- endovenosa (IVC): introdotta nei primi anni ’90, la chemioterapia endovenosa rimane uno dei trattamenti essenziali per il retinoblastoma. L’IVC di solito prevede che due o più agenti chemioterapici vengano somministrati mensilmente attraverso un catetere centrale o periferico per un totale di 6-9 cicli consecutivi. Il regime più comunemente utilizzato è costituito dall’associazione tra vincristina, etoposide e carboplatino (VEC). Dal momento che a seguito del trattamento chemioterapico spesso si osserva una considerevole riduzione delle dimensioni del tumore, l’IVC viene talvolta definita “chemiodulazione”. Le attuali indicazioni per effettuare l’IVC sono: retinoblastoma bilaterale, mutazione germinale confermata, storia familiare di retinoblastoma, casi con sospetta invasione del nervo ottico o della coroide
- intra-arteriosa (IAC): la chemioterapia intra-arteriosa rappresenta una procedura estremamente delicata e costosa che prevede la somministrazione diretta al tumore del chemioterapico attraverso le arterie oftalmiche. Di solito uno, due o addirittura tre farmaci vengono somministrati una volta al mese per una media di tre cicli consecutivi. Questa procedura permette di somministrare direttamente al tumore una dose di chemioterapico 10 volte superiore rispetto a quello che si ottiene con la chemioterapia endovenosa. Negli stadi B e C, in accordo alla classificazione ICRB, di solito è sufficiente una singola dose di farmaco con 5 mg di melfalan per via intra-arteriosa. Tuttavia, una malattia più avanzata con una vasta disseminazione intravitreale o sottoretinica, come accade negli stadi D ed E, o nei casi di tumori refrattari alla terapia, può richiedere una dose maggiore o l’aggiunta di altri chemioterapici come topotecan o carboplatino. Le attuali indicazioni per l’utilizzo dell’IAC sono: terapia di prima scelta nel retinoblastoma unilaterale gruppo B, C, D o E; terapia adiuvante a seguito dell’enucleazione. Nonostante il chemioterapico venga somministrato localmente, la IAC non è risultata scevra da effetti collaterali sistemici quali la neutropenia transitoria o l’occlusione dell’arteria femorale. Gli effetti collaterali oculari possono comprendere edema periorbitale, iperemia cutanea perioculare, madarosi, blefaroptosi e dismotilità extraoculare. Il distacco retinico regmatogeno, probabilmente secondario alla rapida regressione del tumore nel caso di neoplasie a crescita endofitica, è stato riportato nell’8-16% dei casi trattati con IAC.
- intra-oculare per via intravitreale (IVITC): introdotta per la prima volta da Kaneko e Suzuki nel 2003, è risultata utile in combinazione con la chemioterapia intra-arteriosa in quei casi di tumori con disseminazione intra-vitreale o tendenza alla recidiva a seguito di altri trattamenti. La chemioterapia intravitreale non è mai un trattamento di prima linea, ma viene principalmente usata come terapia di salvataggio del globo oculare in caso di fallimento di altre terapie. I farmaci più comunemente usati in IVITC sono il melfalan e il topotecan, da soli o in combinazione. La paracentesi della camera anteriore viene eseguita prima dell’iniezione intravitreale, mentre, dopo l’intravitreale di chemioterapico, viene eseguita una crioterapia a triplo congelamento nel sito d’ngresso dell’ago. Sono stati riportati eventi avversi oculari quali cataratta, emorragia vitreale e/o sottoretinica, ipotonia oculare, ptisi del bulbo, chemosi congiuntivale, eterocromia dell’iride, formazione di sinechie posteriori, uveite anteriore, edema del disco ottico e necrosi retinica emorragica.
- intra-oculare per via intra-camerale (IcamC): introdotta da Munier e colleghi nel 2017, questa modalità di chemioterapia intra-oculare è stata pensata per garantire una maggior disponibilità di chemioterapico in camera anteriore in quei casi di disseminazione anteriore del retinoblastoma, dove in origine era indicata l’enucleazione come terapia di prima linea. La tecnica prevede la somministrazione orale di acetazolamide alla dose di 5 mg/kg prima dell’iniezione, ai fini di inibilire la produzione di umor acqueo e, di conseguenza, ai fini di evitare la diluizione del farmaco. Successivamente, con un approccio trans-corneale, l’umor acqueo viene aspirato e successivamente sostituito con un volume comparabile di melfalan (15-20 μg/0,05 ml) o topotecan (7,5 μg/0,015 ml).
Terapie focali: sono spesso utilizzate per consolidare il trattamento tumorale o in combinazione con IVC o IAC. Le terapie focali attualmente utilizzate comprendono principalmente la crioterapia e la termoterapia transpupillare (TTT), già discusse nel capitolo dedicato al melanoma uveale. Indipendentemente dalla scelta, tutte le terapie focali possono provocare la comparsa di cicatrici corio-retiniche con riduzione del campo visivo e/o dell’acuità visiva, specialmente nel caso di lesioni che si collochino in corrispondenza del polo posteriore.
Radioterapia esterna: prima dell’introduzione della chemioterapia endovenosa, la radioterapia a fascio esterno (EBRT) rappresentava la prima linea di trattamento del retinoblastoma. Oggi l’EBRT, a causa dei numerosi effetti collaterali associati, ha più solo un significato storico. Essa trova ancora indicazione nei casi di estensione extraoculare del tumore, di recidiva orbitaria e di riscontro di margine positivo del nervo ottico a seguito di enucleazione.
Brachiterapia: la brachiterapia o “radioterapia con placca” oggigiorno è usata come trattamento di seconda scelta in caso di tumori chemioresistenti con diametro basale ≤16 mm e spessore compreso tra i 3 e i 9 mm, con o senza disseminazione endovitreale o sottoretinica, a seguito di recidiva dopo IVC o IAC. Lo iodio-125 è l’isotopo più comunemente usato e la dose da fornire all’apice tumorale deve arrivare a 35-40 Gy.
Enucleazione: nonostante i grandi progressi nella gestione del retinoblastoma, l’enucleazione del globo oculare è un trattamento ancora utilizzato. Di solito è riservato a tumori di gruppo E di grandi dimensioni associati a emorragia intravitreale, presenza di estensione extraoculare, sospetta invasione del nervo ottico o della coroide o in caso di tumori in cui sono fallite le pregresse terapie di salvataggio del globo oculare.
Consulenza genetica
La consulenza genetica è un processo che ha come obiettivo quello di prendere in carico il paziente che presenta la patologia che ha basi genetiche (il retinoblastoma) e i familiari a rischio di sviluppare tale patologia.
Il ruolo del consulente genetista risulta duplice: in primo luogo, deve valutare il rischio del probando (in genetica indica il primo individuo esaminato di una famiglia, in cui si riscontra un determinato carattere, e attraverso il quale, si risale alla famiglia portatrice di tale carattere) e dei suoi familiari per lo sviluppo della patologia e guidarli attraverso la serie di test genetici necessari a individuare la/le mutazione/i genetiche responsabili; in secondo luogo dovrà comunicare il risultato di tali test alla famiglia e pianificare il trattamento adeguato per i suoi membri.
Nel caso specifico di un paziente affetto da retinoblastoma, si tratterà di:
- Effettuare un’accurata anamnesi familiare e definire il pedigree
- Identificare le mutazioni responsabili dello sviluppo del retinoblastoma
- Identificare gli individui della famiglia a rischio di sviluppare il retinoblastoma
- Introdurre il paziente in un percorso di cura personalizzato
- Introdurre i familiari a rischio in un percorso di follow up al fine di individuare quanto prima la patologia, qualora questa dovesse svilupparsi.
Ancona-Lezama D, Dalvin LA, Shields CL. Modern treatment of retinoblastoma: A 2020 review. Indian J Ophthalmol. 2020 Nov;68(11):2356-2365. doi: 10.4103/ijo.IJO_721_20. PMID: 33120616; PMCID: PMC7774148.
Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, Gallie BL. Retinoblastoma. Nat Rev Dis Primers. 2015 Aug 27;1:15021. doi: 10.1038/nrdp.2015.21. PMID: 27189421; PMCID: PMC5744255.
Bouchoucha Y, Matet A, Berger A, Carcaboso AM, Gerrish A, Moll A, Jenkinson H, Ketteler P, Dorsman JC, Chantada G, Beck-Popovic M, Munier F, Aerts I, Doz F, Golmard L; European Retinoblastoma Group EuRbG. Retinoblastoma: From genes to patient care. Eur J Med Genet. 2023 Jan;66(1):104674. doi: 10.1016/j.ejmg.2022.104674. Epub 2022 Dec 5. PMID: 36470558.
Soliman SE, Racher H, Zhang C, MacDonald H, Gallie BL. Genetics and Molecular Diagnostics in Retinoblastoma–An Update. Asia Pac J Ophthalmol (Phila). 2017 Mar-Apr;6(2):197-207. doi: 10.22608/APO.201711. PMID: 28399338.
Arun D. Singh, A. Linn Murphree, Bertil E. Damato. Clinical Ophthalmic Oncology, Retinoblastoma.
Global Retinoblastoma Study Group. The Global Retinoblastoma Outcome Study: a prospective, cluster-based analysis of 4064 patients from 149 countries. Lancet Glob Health. 2022 Aug;10(8):e1128-e1140. doi: 10.1016/S2214-109X(22)00250-9. PMID: 35839812; PMCID: PMC9397647.
